A Doença de Wilson é uma doença genética rara caracterizada pelo acúmulo excessivo de cobre no organismo, principalmente no fígado, sistema nervoso central e outros órgãos. Quando não tratada, pode evoluir para cirrose, insuficiência hepática ou complicações neurológicas graves.
A Doença de Wilson é causada por mutações no gene ATP7B. A condição segue um padrão autossômico recessivo, ou seja, é necessário que o indivíduo herde duas cópias alteradas do gene (uma de cada genitor) para manifestar a doença.
Sintomas
Os sintomas da doença variam de acordo com o órgão mais afetado e a idade do paciente. Entre os principais sintomas estão:
- Sintomas hepáticos: icterícia, hepatomegalia, fadiga, náuseas e cirrose progressiva
- Sintomas neurológicos: tremores, rigidez muscular, alteração na coordenação motora, alterações na fala, alterações de marcha
- Sintomas psiquiátricos: irritabilidade, depressão e ansiedade
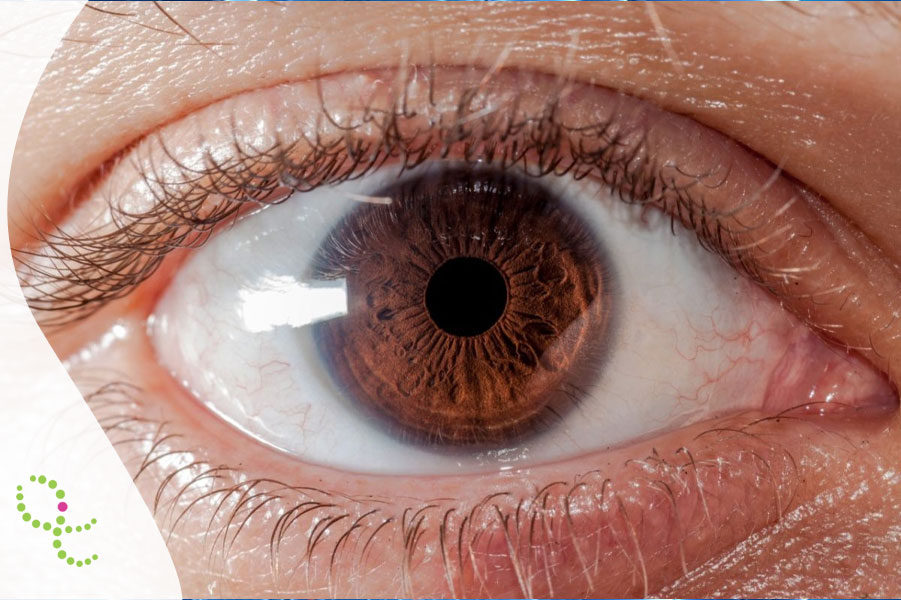
- Sintomas oftalmológicos: anel de Kayser-Fleischer (depósito de cobre na córnea)
Diagnóstico
O diagnóstico da Doença de Wilson é feito com base na combinação de anamnese, exame físico (a presença do anel de Kayser-Fleischer é um achado importante), exames laboratoriais (ceruloplasmina sérica diminuída e cobre urinário de 24 horas aumentado) e exames de imagem. Os testes genéticos para mutações no gene ATP7B podem confirmar o diagnóstico, especialmente em casos familiares ou atípicos.
Tratamento
O tratamento da doença de Wilson tem como objetivo reduzir o acúmulo de cobre no organismo e prevenir danos ao fígado e ao sistema nervoso. Isso é feito principalmente com medicações quelantes, como a penicilamina ou trientina, que ajudam o corpo a eliminar o cobre, e com suplementos de zinco, que diminuem a absorção de cobre pelo intestino. Além disso, é importante seguir uma dieta com restrição de alimentos ricos em cobre. Em casos graves de comprometimento hepático, pode ser necessário transplante de fígado.








